| Research |
|
§ Atomistic structure prediction of materials has been a central focus on physical, chemical, and materials sciences, but it is extremely difficult as it basically involves in classifying a huge number of energy minima on the multi-dimensional lattice energy surface. We focus on the development of global optimization methods on structure prediction with the only given information of chemical compositions for materials at certain external conditions (e.g., temperature and pressure). An efficient CALYPSO (Crystal structural AnLYsis by Particle Swarm Optimization) approach for structure prediction based on swarm intelligence algorithm by taking the advantage of structures smart learning was developed and coded into the CALYPSO software (http://www.calypso.cn) which is free for academic use. Currently, CALYPSO software is able to predict structures of three-dimensional crystals, isolated nano-clusters or molecules, surface reconstructions, two-dimensional layers, and functionality-driven design of superhard and optical materials. |
§ Pressure is one of the fundamental thermodynamic variables, which can allow precisely tuning of the inter-atomic distance and alter the bonding patterns of materials, leading to phase transformations into high-pressure structures with unusual chemical and physical properties. High-pressure structure investigations based on CALYPSO structure searching technique in conjunction with first-principles calculations are extensively carried out in our group in an effort to explore exotic crystal structures, new physics and chemistry of materials under high pressure. In-situ high-pressure experiments via the guidance of our theory might be also performed in our group or in some cases in collaboration with other groups to verify our theoretical predictions. |
§ Materials design has been the subject of topical interests in materials and physical sciences. Atomistic structures of materials occupy a central and often critical role, when establishing a correspondence between materials performance and their basic compositions. Structural design of novel functional materials (e.g., superhard, thermoelectric, superconductive, magnetic, optical, and high-energy materials) through CALYPSO structure prediction and first-principles calculations is one of the main research directions in our group in an effort to accelerate materials discovery. Besides for some efforts at ambient pressure, high pressure is often used as an efficient tool in our research to discover exotic materials, which are not accessible at ambient conditions. Experimental synthesis with the theoretical guidance might be also performed in our group or in some cases in collaboration with other groups. |
| Highlights |
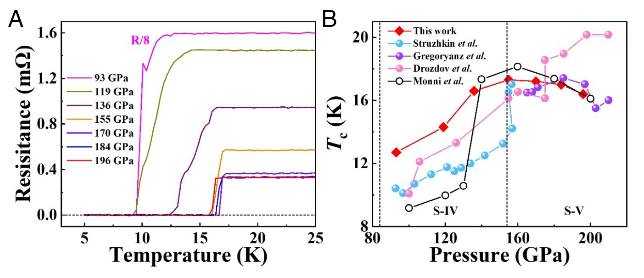
The abrupt drop of resistance to zero at a critical temperature is a key signature of the current paradigm of the metal–superconductor transition. However, the emergence of an intermediate bosonic insulating state characterized by a resistance peak preceding the onset of the superconducting transition has challenged this traditional understanding. Notably, this phenomenon has been predominantly observed in disordered or chemically doped low-dimensional systems, raising intriguing questions about the generality of the effect and its underlying fundamental physics. Here, we present a systematic experimental study of compressed elemental sulfur, an undoped three-dimensional (3D) high-pressure superconductor, with detailed measurements of electrical resistance as a function of tem- perature, magnetic field, and current. The anomalous resistance peak observed in this 3D system is interpreted based on an empirical model of a metal–bosonic insulator–supercon- ductor transition, potentially driven by vortex dynamics under magnetic field and energy dissipation processes. These findings offer a fresh platform for theoretical analysis of the decades-long enigmatic of the underlying mechanism of this phenomenon. The research results have been published in Proc. Natl. Acad. Sci. USA 122 , e2420904122 (2025) |
 |
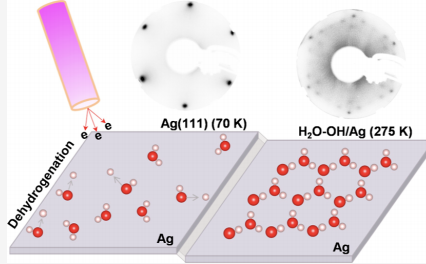
Water adsorption on metal surfaces is ubiquitous in broad natural and technological settings. However, elucidating this phenomenon is often challenging due to difficulties in accurately determining the morphology and understanding the chemistry of adsorbed water networks. Here, we report a significant discovery of the water-hydroxyl (H2O–OH) wetting monolayer, which has long been deemed possible only on hydrophilic metal surfaces, now realized on an archetypal hydrophobic metal surface, Ag(111). Ab initio structure searches predicted a hexagonal hydrogen-bonded network comprising alternating H2O and OH units; ensuing low-energy-electron-assisted synthesis in concert with extensive characterization and computational simulation provided compelling evidence of an H2O–OH monolayer realized on the Ag(111) surface, with remarkable stability up to near room temperature. Our finding brings new insights into the intriguing chemistry of H2O–OH overlayers on metal surfaces, and the electron-assisted synthesis opens a unique pathway toward creating delicate molecular networks. The research results have been published in J. Am. Chem. Soc. 147, 21162 (2025) |
 |
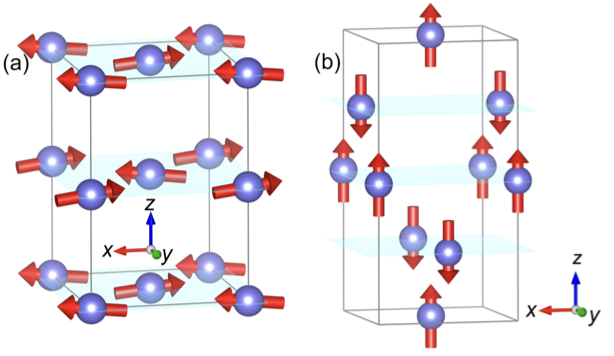
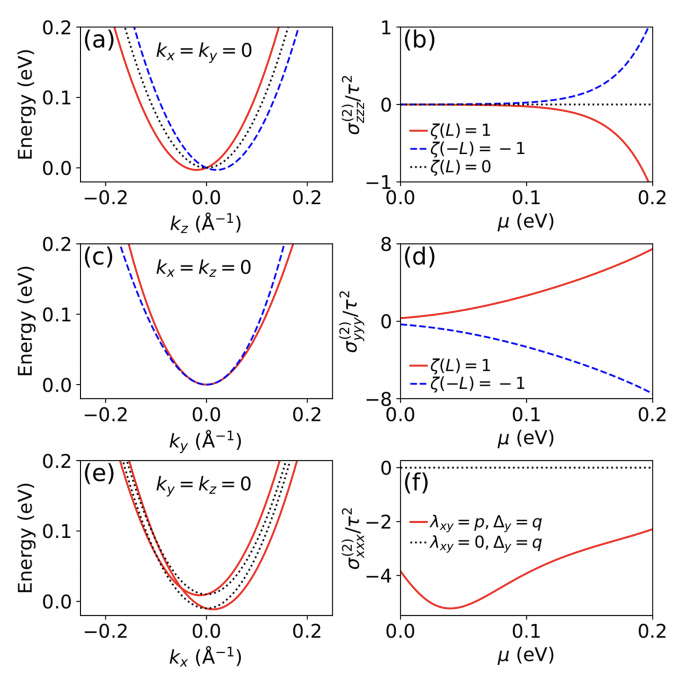
Writing data by electric field (as opposed to electric current) offers promises for energy efficient memory devices. While this data writing scheme is enabled by the magnetoelectric effect, the narrow spectrum of room-temperature magnetoelectrics hinders the design of practical magnetoelectric memories, and the exploration of other mechanisms toward low-power memories is greatly demanding. Here, we propose a mechanism that allows the electric-field writing of data beyond the framework of magnetoelectric effect. By symmetry analysis, we show that electric field can induce longitudinal nonlinear conductivity (LNC) in a wide spectrum of magnetic materials, including ferromagnets, antiferromagnets, magnetoelectrics, and nonmagnetoelectrics. The LNC is electrically switchable by reversing the electric field, where the switched LNC is detectable by transport measurements. Our first-principles simulations combined with transport calculations further predict YFeO3 and CuFeS2 (room-temperature antiferromagnets) to showcase electrically switchable LNC. Our Letter helps enrich the research avenues in nonlinear charge transport, and offers a pathway for designing energy efficient devices based on LNC. The research results have been published in Phys. Rev. Lett. 134 , 046801 (2025) |
 |
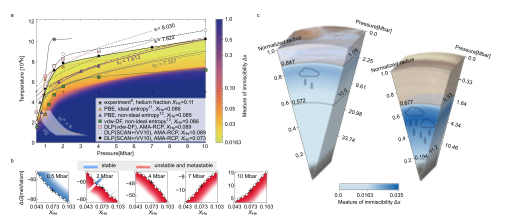
The immiscibility of hydrogen-helium mixture under the temperature and pressure conditions of planetary interiors is crucial for understanding the structures of gas giant planets (e.g., Jupiter and Saturn). While the experimental probe at such extreme conditions is challenging, theoretical simulation is heavily relied in an effort to unravel the mixing behavior of hydrogen and helium. Here we develop a method via a machine learning accelerated molecular dynamics simulation to quantify the physical separation of hydrogen and helium under the conditions of planetary interiors. The immiscibility line achieved with the developed method yields substantially higher demixing temperatures at pressure above 1.5 Mbar than earlier theoretical data, but matches better to the experimental estimate. Our results suggest a possibility that H-He demixing takes place in a large fraction of the interior radii of Jupiter and Saturn, i.e., 27.5% in Jupiter and 48.3% in Saturn. This indication of an H-He immiscible layer hints at the formation of helium rain and offers a potential explanation for the decrease of helium in the atmospheres of Jupiter and Saturn. The research results have been published in Nature Commun. 15, 8543 (2024) |
 |
Room-temperature superconductivity has been a long-held dream of condensed matter physics for over a century. Here, we predict a class of thermodynamically stable clathrate hydrides consisting of two previously unreported H24 and H30 cages at megabar pressures. Among these hydrides, LaSc2H24 is calculated to exhibit superconductivity with Tc up to 316 K at 167 GPa even when considering the effects of anharmonicity. Such “hot” superconductivity above room temperature is attributed to an unusually large hydrogen-derived density of states at the Fermi level arising from the unusual combination of H30 and H24 cages of this structure. Further exploration of the La–Sc–H ternary system is expected to facilitate the creation of other high-temperature superconductors. The research results have been published in Proc. Natl. Acad. Sci. USA 121, e2401840121 (2024) |
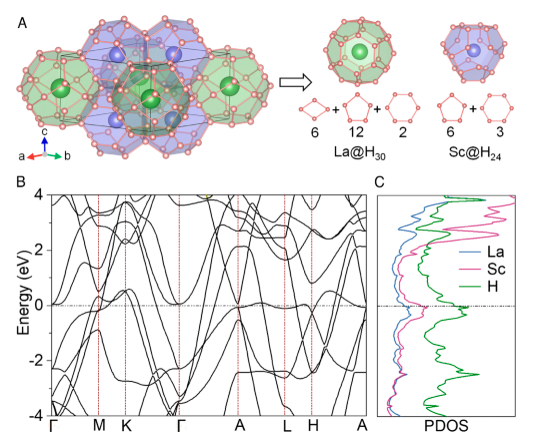 |
The metal–bosonic insulator–superconductor transition, predominantly observed in low-dimensional systems with disorder or magnetic impurities, defies mean-field theory and challenges the long-held picture of a straightforward transition from metal to either superconductor or insulator. Here, we report experimental evidence of a magnetic field-tuned bosonic insulating state in compressed 3D sulfur in the vicinity of its superconducting transition. The experiments reveal a prominent concurrent resistance peak as a function of temperature, magnetic field, and current in the same superconductor. The results suggest the potential for revealing diverse electronic and transport phenomena in putatively simple systems and open avenues for advancing the understanding of the underlying physics. The research results have been published in Proc. Natl. Acad. Sci. USA 122, e2420904122 (2024) |
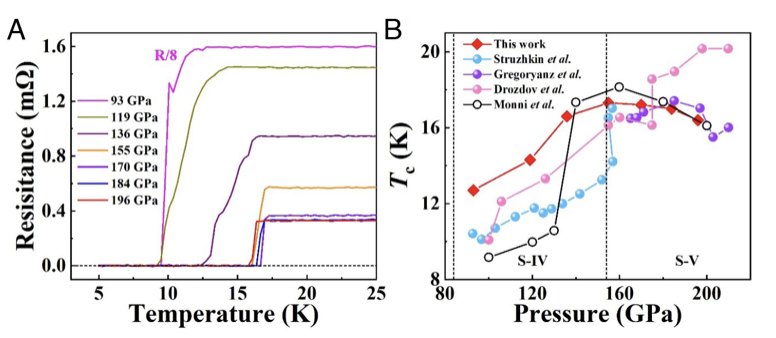 |
Ferroelectricity in CMOS-compatible hafnia (HfO2) is crucial for the fabrication of high-integration nonvolatile memory devices. However, the capture of ferroelectricity in HfO2 requires the stabilization of thermodynamically metastable orthorhombic or rhombohedral phases, which entails the introduction of defects (e.g., dopants and vacancies) and pays the price of crystal imperfections, causing unpleasant wake-up and fatigue effects. Here, we report a theoretical strategy on the realization of robust ferroelectricity in HfO2-based ferroelectrics by designing a series of epitaxial (HfO2)1/(CeO2)1 superlattices. The designed ferroelectric superlattices are defects free, and most importantly, on the base of the thermodynamically stable monoclinic phase of HfO2. Consequently, this allows the creation of superior ferroelectric properties with an electric polarization >25 μC/cm2 and an ultralow polarization-switching energy barrier at ∼2.5 meV/atom. Our work may open an avenue toward the fabrication of high-performance HfO2-based ferroelectric devices. The research results have been published in Phys. Rev. Lett. 132, 256801 (2024) |
 |
Dense hydrous magnesium silicate MgSiO4H2 is widely regarded as a primary water carrier into the deep Earth. However, the stability fields of MgSiO4H2 based on the prevailing structure model are narrower than experimental results at relevant pressure and temperature (P-T) conditions, casting doubts about this prominent mineral as a water carrier into the great depths of the Earth. Here, we report on an advanced structure search that identifies two new crystal structures, denoted as 𝛼- and 𝛽−MgSiO4H2, that are stable over unprecedentedly wide P-T conditions of 17–68 GPa and up to 1860 K, covering the entire experimentally determined range. Moreover, we performed x-ray diffraction measurements with backscattering electron image, combined with ab initio simulations, to demonstrate the formation of MgSiO4H2 and AlOOH solid solutions that exhibit further enhanced P-T stability fields, making them robust carriers of water into the deepest lower mantle. These findings establish and elucidate the new MgSiO4H2 phases as potential primary water carriers into the vast depths of the lower mantle, creating a distinct paradigm for the deep Earth water cycle. The research results have been published in Phys. Rev. Lett. 133, 214101 (2024) |
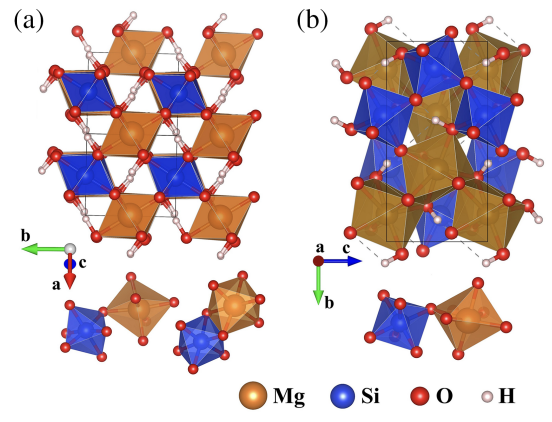 |
The longitudinal nonreciprocal charge transport (NCT) in crystalline materials is a highly nontrivial phenomenon, motivating the design of next generation two-terminal rectification devices (e.g., semiconductor diodes beyond PN junctions). The practical application of such devices is built upon crystalline materials whose longitudinal NCT occurs at room temperature and under low magnetic field. However, materials of this type are rather rare and elusive, and theory guiding the discovery of these materials is lacking. Here, we develop such a theory within the framework of semiclassical Boltzmann transport theory. By symmetry analysis, we classify the complete 122 magnetic point groups with respect to the longitudinal NCT phenomenon. The symmetry-adapted Hamiltonian analysis further uncovers a previously overlooked mechanism for this phenomenon. Our theory guides the first-principles prediction of longitudinal NCT in multiferroic 𝜖−Fe2O3 semiconductor that possibly occurs at room temperature, without the application of external magnetic field. These findings advance our fundamental understandings of longitudinal NCT in crystalline materials, and aid the corresponding materials discoveries. The research results have been published in Phys. Rev. Lett. 133, 096802 (2024) |
 |
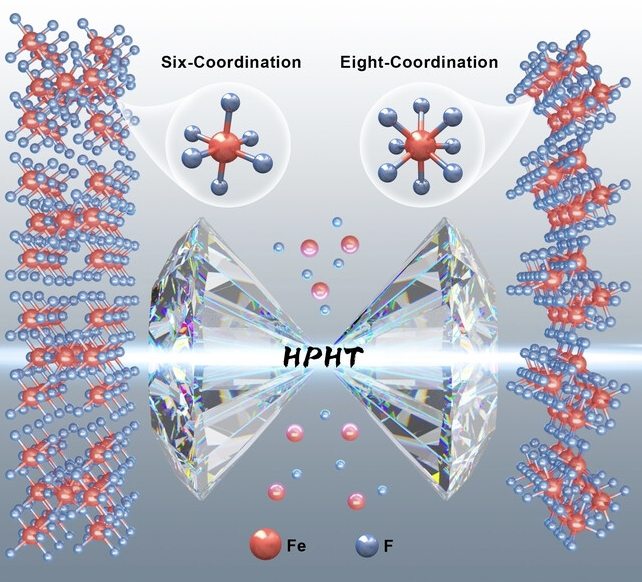
The chemistry of hypercoordination has been a subject of fundamental interest, especially for understanding structures that challenge conventional wisdom. The small ionic radii of Fe ions typically result in coordination numbers of 4 or 6 in stable Fe-bearing ionic compounds. While 8-coordinated Fe has been observed in highly compressed oxides, the pursuit of hypercoordinated Fe still faces significant challenges due to the complexity of synthesizing the anticipated compound with another suitable anion. Through first-principles simulation and advanced crystal structure prediction methods, we predict that an orthorhombic phase of FeF3 with exclusively 8-coordinated Fe is energetically stable above 18 GPa—a pressure more feasibly achieved compared to oxides. Inspired by this theoretical result, we conducted extensive experiments using a laser-heated diamond anvil cell technique to investigate the crystal structures of FeF3 at high-pressure conditions. We successfully synthesized the predicted orthorhombic phase of FeF3 at 46 GPa, as confirmed by in situ experimental X-ray diffraction data. This work establishes a new ionic compound featuring rare 8-coordinated Fe in a simple binary Fe-bearing system and paves the way for discovering Fe hypercoordination in similar systems. The research results have been published in Angew. Chem.Int. Ed. 63, e2023193 (2024) |
 |
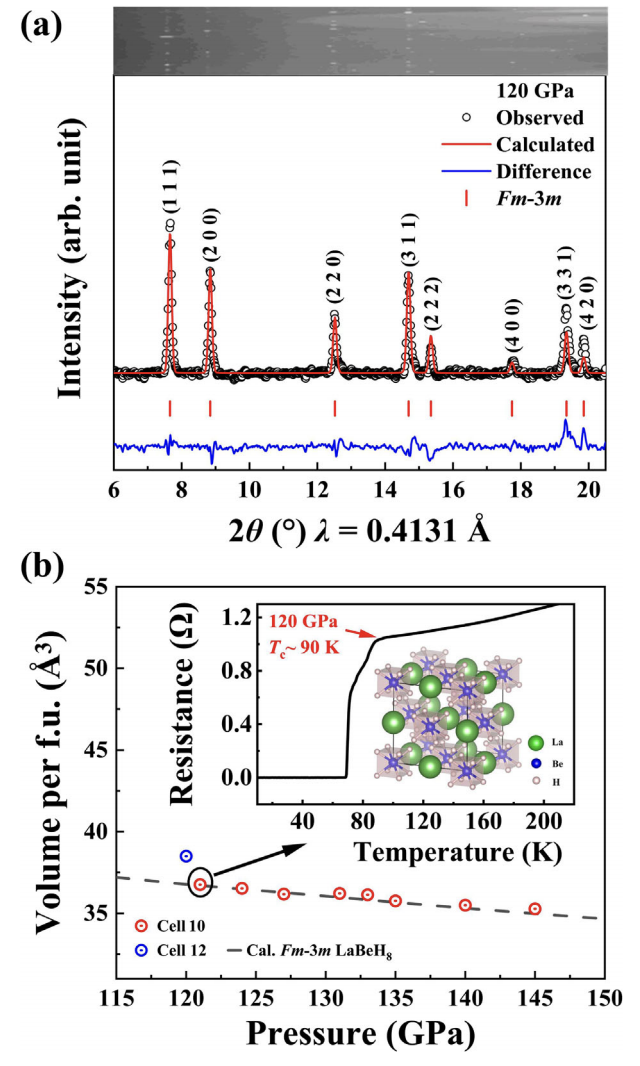
The search for high-temperature superconducting superhydrides has recently moved into a new phase by going beyond extensively probed binary compounds and focusing on ternary ones with vastly expanded material types and configurations for property optimization. Theoretical and experimental works have revealed promising ternary compounds that superconduct at or above room temperature, but it remains a pressing challenge to synthesize stoichiometric ternary compounds with a well-resolved crystal structure that can host high-temperature superconductivity at submegabar pressures. Here, we report on the successful synthesis of ternary LaBeH8 obtained via compression in a diamond anvil cell under 110–130 GPa. X-ray diffraction unveils a rocksalt-like structure composing La and BeH8 units in the lattice. Transport measurements determined superconductivity with critical temperature 𝑇𝑐 up to 110 K at 80 GPa, as evidenced by a sharp drop of resistivity to zero and a characteristic shift of 𝑇𝑐 driven by a magnetic field. Our experiment establishes the first superconductive ternary compound with a resolved crystal structure. These findings raise the prospects of rational development of the class of high-𝑇𝑐 superhydrides among ternary compounds, opening greatly expanded and more diverse structural space for exploration and discovery of superhydrides with enhanced high-𝑇𝑐 superconductivity. The research results have been published in Phys. Rev. Lett. 130 , 266001 (2023) |
 |
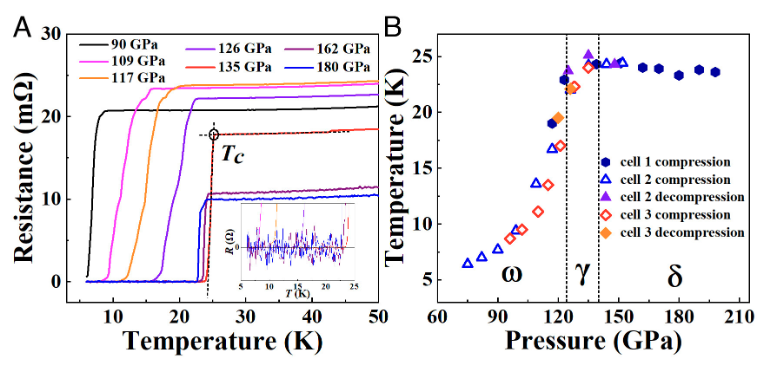
The anomalous metallic state (AMS) emerging from a quantum superconductor-to-metal transition is a subject of great current interest since this exotic quantum state exhibits unconventional transport properties that challenge the core physics principles of Fermi liquid theory. As the AMS concept is historically derived from disordered two-dimensional (2D) systems, related studies have predominately concentrated on 2D materials. The AMS behaviors in three-dimensional (3D) systems have been rarely reported to date, which raises intriguing questions on the fundamental nature of pertinent physics. Here, we report experimental evidence for a 3D AMS in highly compressed titanium metal that exhibits superconductivity with a critical temperature (Tc) reaching near-record 25.1 K among elemental superconductors, offering a favorable material template for exploring 3D AMS. At sufficiently strong magnetic fields, unusual transport behaviors set in over a wide pressure range, showcasing AMS hallmarks of a low-temperature saturation resistance below the Drude value and giant positive magnetoresistance. These findings reveal a 3D AMS in simple elemental systems and, more importantly, provide a fresh platform for probing the decades-long enigmatic underlying physics. The research results have been published in Proc. Natl. Acad. Sci. USA 120 , e2218856120 (2023) |
 |
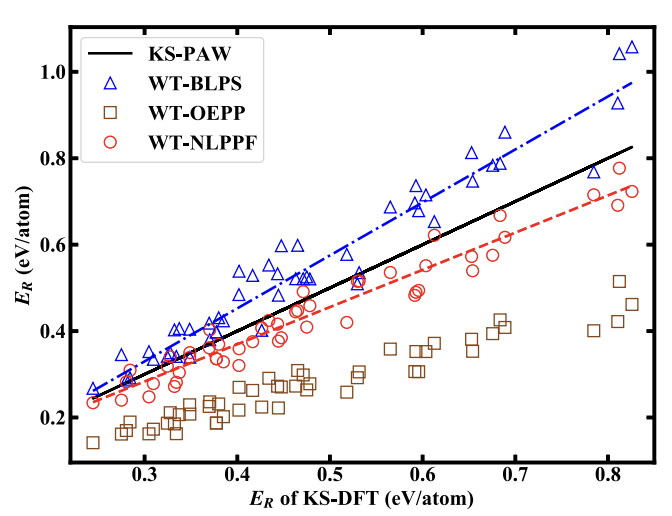
Orbital-free density functional theory (OF-DFT) is an electronic structure method with a low computational cost that scales linearly with the number of simulated atoms, making it suitable for large-scale material simulations. It is generally considered that OF-DFT strictly requires the use of local pseudopotentials, rather than orbital-dependent nonlocal pseudopotentials, for the calculation of electron-ion interaction energies, as no orbitals are available. This is unfortunate situation since the nonlocal pseudopotentials are known to give much better transferability and calculation accuracy than local ones. We report here the development of a theoretical scheme that allows the direct use of nonlocal pseudopotentials in OF-DFT. In this scheme, a nonlocal pseudopotential energy density functional is derived by the projection of nonlocal pseudopotential onto the non-interacting density matrix (instead of “orbitals”) that can be approximated explicitly as a functional of electron density. Our development defies the belief that nonlocal pseudopotentials are not applicable to OF-DFT, leading to the creation for an alternate theoretical framework of OF-DFT that works superior to the traditional approach. The research results have been published in Nature Commun. 13 , 1385 (2022) |
 |
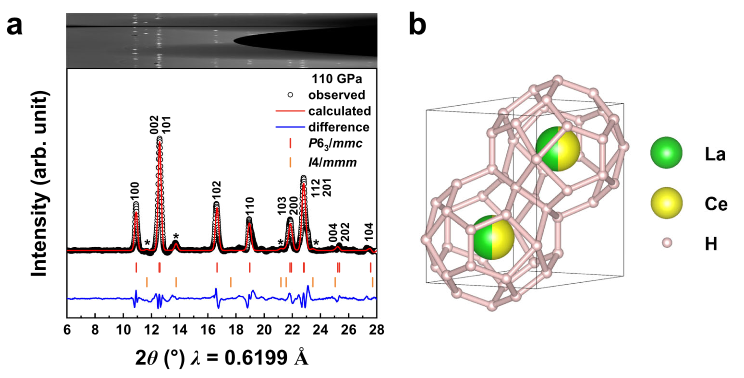
A sharp focus of current research on superconducting superhydrides is to raise their critical temperature Tc at moderate pressures. Here, we report a discovery of giant enhancement of Tc in CeH9 obtained via random substitution of half Ce by La, leading to equal-atomic (La,Ce)H9 alloy stabilized by maximum configurational entropy, containing the LaH9 unit that is unstable in pure compound form. The synthesized (La,Ce)H9 alloy exhibits Tc of 148–178 K in the pressure range of 97–172 GPa, representing up to 80% enhancement of Tc compared to pure CeH9 and showcasing the highest Tc at sub-megabar pressure among the known superhydrides. This work demonstrates substitutional alloying as a highly effective enabling tool for substantially enhancing Tc via atypical compositional modulation inside suitably selected host crystal. This optimal substitutional alloying approach opens a promising avenue for synthesis of high-entropy multinary superhydrides that may exhibit further increased Tc at even lower pressures. The research results have been published in Nature Commun. 13 , 5925 (2022) |
 |
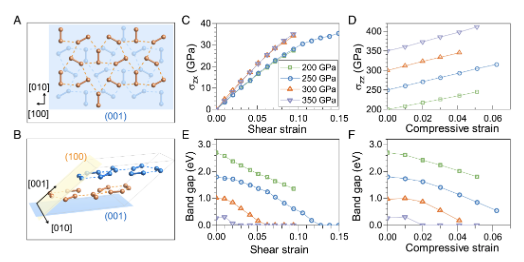
Solid molecular hydrogen has been predicted to be metallic and high-temperature superconducting at ultrahigh hydrostatic pressures that push current experimental limits. Meanwhile, little is known about the influence of nonhydrostatic conditions on its electronic properties at extreme pressures where anisotropic stresses are inevitably present and may also be intentionally introduced. Here we show by first-principles calculations that solid molecular hydrogen compressed to multimegabar pressures can sustain large anisotropic compressive or shear stresses that, in turn, cause major crystal symmetry reduction and charge redistribution that accelerate bandgap closure and promote superconductivity relative to pure hydrostatic compression. Our findings highlight a hitherto largely unexplored mechanism for creating superconducting dense hydrogen, with implications for exploring similar phenomena in hydrogen-rich compounds and other molecular crystals. The research results have been published in Proc. Natl. Acad. Sci. USA 119 , e2122691119 (2022) |
 |
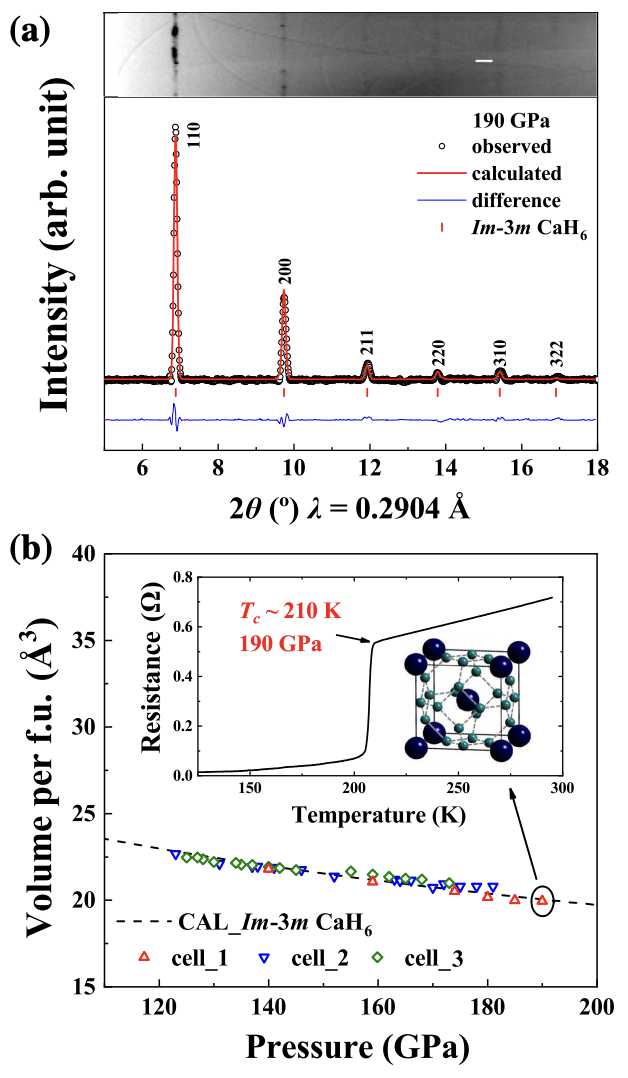
The recent discovery of superconductive rare earth and actinide superhydrides has ushered in a new era of superconductivity research at high pressures. This distinct type of clathrate metal hydrides was first proposed for alkaline-earth-metal hydride CaH6 that, however, has long eluded experimental synthesis, impeding an understanding of pertinent physics. Here, we report successful synthesis of CaH6 and its measured superconducting critical temperature 𝑇𝑐 of 215 K at 172 GPa, which is evidenced by a sharp drop of resistivity to zero and a characteristic decrease of 𝑇𝑐 under a magnetic field up to 9 T. An estimate based on the Werthamer-Helfand-Hohenberg model gives a giant zero-temperature upper critical magnetic field of 203 T. These remarkable benchmark superconducting properties place CaH6 among the most outstanding high-𝑇𝑐 superhydrides, marking it as the hitherto only clathrate metal hydride outside the family of rare earth and actinide hydrides. This exceptional case raises great prospects of expanding the extraordinary class of high-𝑇𝑐 superhydrides to a broader variety of compounds that possess more diverse material features and physics characteristics. The research results have been published in Phys. Rev. Lett. 128 , 167001 (2022) |
 |
Materials once suffered at high-pressure and high-temperature (HPHT) conditions often exhibit exotic phenomena that defy conventional wisdom. The behaviors of sulfur dioxide (SO2), one of the archetypal simple molecules, at HPHT conditions have attracted a great deal of attention due to its relevance to the S cycle between deep Earth and the atmosphere. Here we report the discovery of an unexpected disproportionation of SO2 via bond breaking into elemental S and sulfur trioxide (SO3) at HPHT conditions through a jointly experimental and theoretical study. Measured x-ray diffraction and Raman spectroscopy data allow us to solve unambiguously the crystal structure (space group R-3c) of the resultant SO3 phase that shows an extended framework structure formed by vertex-sharing octahedra SO6. Our findings lead to a significant extension of the phase diagram of SO2 and suggest that SO2, despite its abundance in Earth’s atmosphere and ubiquity in other giant planets, is not a stable compound at HPHT conditions relevant to planetary interiors, providing important implications for elucidating the S chemistry in deep Earth and other giant planets. The research results have been published in Phys. Rev. Lett. 128 , 106001 (2022) |
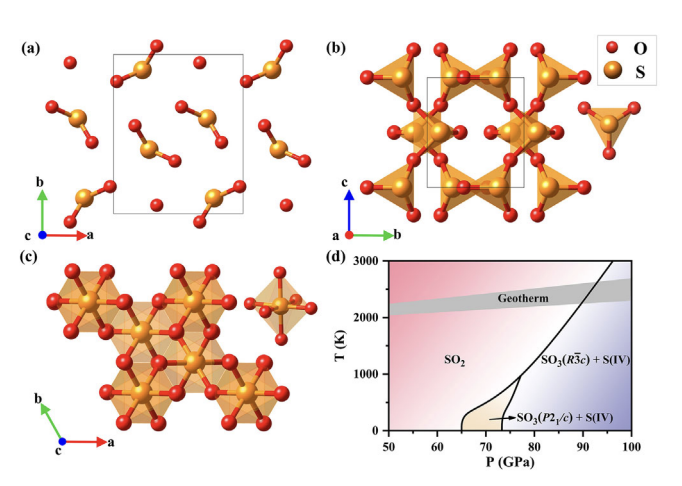 |
Centrosymmetric antiferromagnetic semiconductors, although abundant in nature, seem less promising than ferromagnets and ferroelectrics for practical applications in semiconductor spintronics. As a matter of fact, the lack of spontaneous polarization and magnetization hinders the efficient utilization of electronic spin in these materials. Here, we propose a paradigm to harness electronic spin in centrosymmetric antiferromagnets via Zeeman spin splitting of electronic energy levels—termed as the spin Zeeman effect—which is controlled by an electric field. By symmetry analysis, we identify 21 centrosymmetric magnetic point groups that accommodate such a spin Zeeman effect. We further predict by first principles that two antiferromagnetic semiconductors, Fe2TeO6 and SrFe2S2O, are excellent candidates showcasing Zeeman splittings as large as ∼55 and ∼30 meV, respectively, induced by an electric field of 6 MV/cm. Moreover, the electronic spin magnetization associated to the splitting energy levels can be switched by reversing the electric field. Our Letter thus sheds light on the electric-field control of electronic spin in antiferromagnets, which broadens the scope of application of centrosymmetric antiferromagnetic semiconductors. The research results have been published in Phys. Rev. Lett. 129 , 187602 (2022) |
 |
Materials containing planar hypercoordinate motifs greatly enriched the fundamental understanding of chemical bonding. Herein, by means of first-principles calculations combined with global minimum search, we discovered the two-dimensional (2D) SrB8 monolayer, which has the highest planar coordination number (12) reported so far in extended periodic materials. In the SrB8 monolayer, bridged B8 units are forming the boron monolayer consisting of B12 rings, and the Sr atoms are embedded at the center of these B12 rings, leading to the Sr@B12 motifs. The SrB8 monolayer has good thermodynamic, kinetic, and thermal stabilities, which is attributed to the geometry fit between the size of the Sr atom and cavity of the B12 rings, as well as the electron transfer from Sr atoms to electron-deficient boron network. Placing the SrB8 monolayer on the Ag(001) surface shows good commensurability of the lattices and small vertical structure undulations, suggesting the feasibility of its experimental realization by epitaxial growth. Potential applications of the SrB8 monolayer on metal ions storage (for Li, Na, and K) are explored. The research results have been published in J. Am. Chem. Soc. 144 , 11120 (2022) |
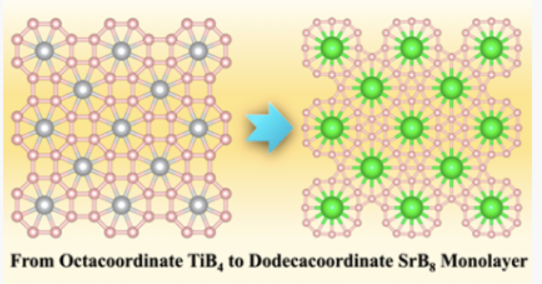 |
Achieving room-temperature superconductivity has been an enduring scientific pursuit driven by broad fundamental interest and enticing potential applications. The recent discovery of high-pressure clathrate superhydride LaH10 with superconducting critical temperatures (Tc) of 250–260 K made it tantalizingly close to realizing this long-sought goal. Here, we report a remarkable finding based on an advanced crystal structure search method of a new class of extremely hydrogen-rich clathrate superhydride MH18 (M: rare-earth/actinide atom) stoichiometric compounds stabilized at an experimentally accessible pressure of 350 GPa. These compounds are predicted to host Tc up to 330 K, which is well above room temperature. The bonding and electronic properties of these MH18 clathrate superhydrides closely resemble those of atomic metallic hydrogen, giving rise to the highest Tc hitherto found in a thermodynamically stable hydride compound. An in-depth study of these extreme superhydrides offers insights for elucidating phonon-mediated superconductivity above room temperature in hydrogen-rich and other low-Z materials. The research results have been published in J. Am. Chem. Soc. 144 , 13394 (2022) |
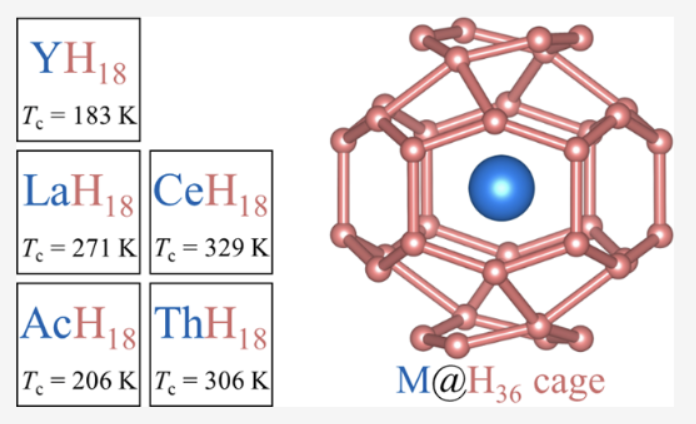 |
Understanding the interior structure and dynamics of outer Solar System planets in terms of their component materials is a major scientific challenge. A highly intriguing case concerns the anomalous nondipolar and nonaxisymmetric magnetic fields of Uranus and Neptune. A thin-shell dynamo model has been shown to capture observed phenomena but leaves unexplained its origin and materials basis. We report extensive theoretical calculations that indicate the stability of trihydrogen oxide (H3O) in solid, superionic, and fluid metallic states at the deep interior conditions of these planets. The fluid metallic phase is stable in a thin-shell zone near their cores and exhibits the properties required to produce the observed enigmatic magnetic fields. These findings have implications for other planets, including related exoplanets. The research results have been published in Proc. Natl. Acad. Sci. USA 117, 5638 (2020) |
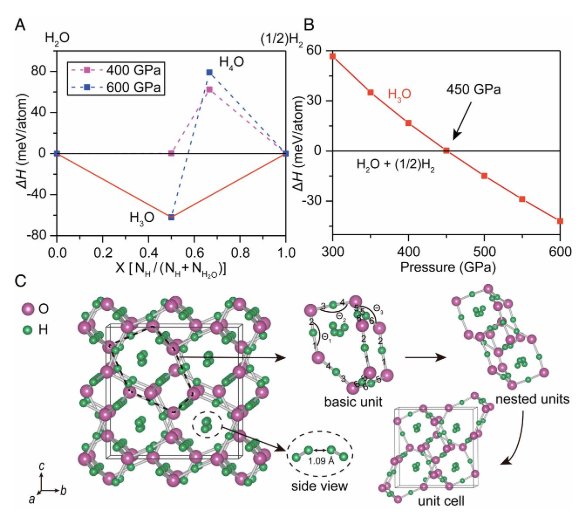 |
Diamond is a prototypical ultrawide band gap semiconductor, but turns into a superconductor with a critical temperature Tc≈4 K near 3% boron doping [E. A. Ekimov et al., Nature (London) 428, 542 (2004)]. Here we unveil a surprising new route to superconductivity in undoped diamond by compression-shear deformation that induces increasing metallization and lattice softening with rising strain, producing phonon mediated Tc up to 2.4–12.4 K for a wide range of Coulomb pseudopotential μ*=0.15–0.05. This finding raises intriguing prospects of generating robust superconductivity in strained diamond crystal, showcasing a distinct and hitherto little explored approach to driving materials into superconducting states via strain engineering. These results hold promise for discovering superconductivity in normally nonsuperconductive materials, thereby expanding the landscape of viable nontraditional superconductors and offering actionable insights for experimental exploration. The research results have been published in Phys. Rev. Lett. 124, 147001 (2020) |
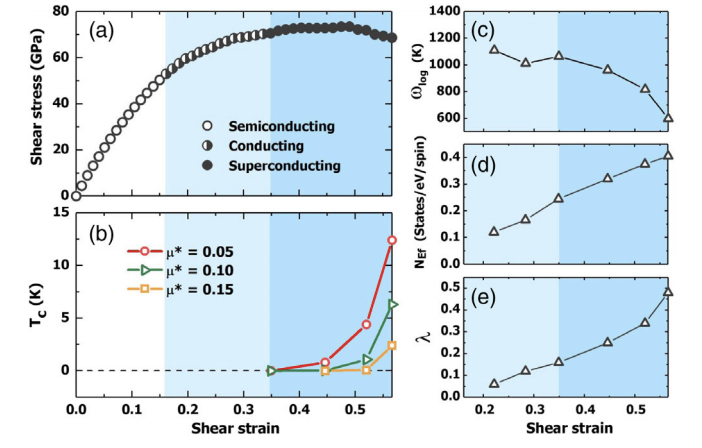 |
High pressure can drastically alter chemical bonding and produce exotic compounds that defy conventional wisdom. Especially significant are compounds pertaining to oxygen cycles inside Earth, which hold key to understanding major geological events that impact the environment essential to life on Earth. Here we report the discovery of pressure-stabilized divalent ozonide CaO3 crystal that exhibits intriguing bonding and oxidation states with profound geological implications. Our computational study identifies a crystalline phase of CaO3 by reaction of CaO and O2 at high pressure and high temperature conditions; ensuing experiments synthesize this rare compound under compression in a diamond anvil cell with laser heating. High-pressure x-ray diffraction data show that CaO3 crystal forms at 35 GPa and persists down to 20 GPa on decompression. Analysis of charge states reveals a formal oxidation state of −2 for ozone anions in CaO3. These findings unravel the ozonide chemistry at high pressure and offer insights for elucidating prominent seismic anomalies and oxygen cycles in Earth’s interior. We further predict multiple reactions producing CaO3 by geologically abundant mineral precursors at various depths in Earth’s mantle. The research results have been published in Phys. Rev. Lett. 123, 195504 (2019) |
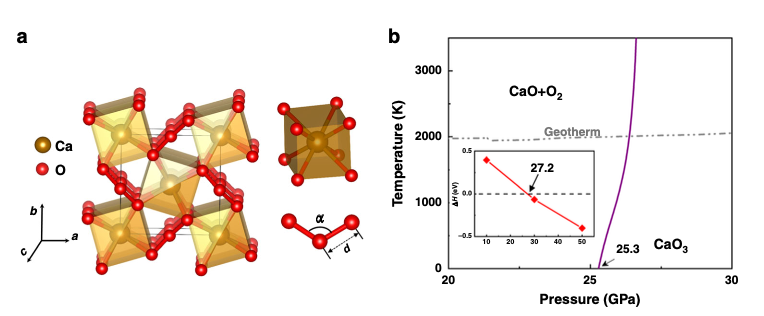 |
Diamond is the quintessential superhard material widely known for its stiff and brittle nature and large electronic band gap. In stark contrast to these established benchmarks, our first-principles studies unveil surprising intrinsic structural ductility and electronic conductivity in diamond under coexisting large shear and compressive strains. These complex loading conditions impede brittle fracture modes and promote atomistic ductility, triggering rare smooth plastic flow in the normally rigid diamond crystal. This extraordinary structural change induces a concomitant band gap closure, enabling smooth charge flow in deformation created conducting channels. These startling soft-and-conducting modes reveal unprecedented fundamental characteristics of diamond, with profound implications for elucidating and predicting diamond’s anomalous behaviors at extreme conditions. The research results have been published in Phys. Rev. Lett. 123, 195504 (2019) |
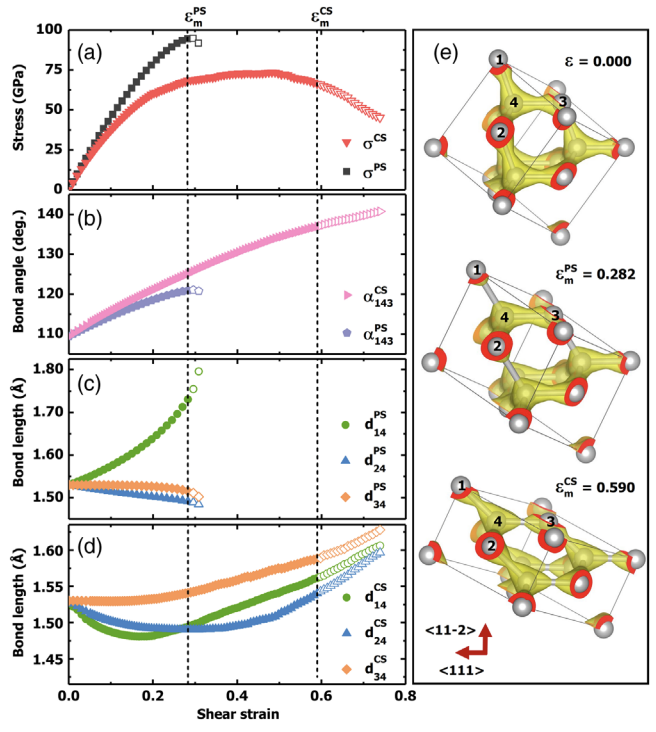 |
The recent theory-orientated discovery of record high-temperature superconductivity (Tc∼250 K) in sodalitelike clathrate LaH10 is an important advance toward room-temperature superconductors. Here, we identify an alternative clathrate structure in ternary Li2MgH16 with a remarkably high estimated Tc of ∼473 K at 250 GPa, which may allow us to obtain room-temperature or even higher-temperature superconductivity. The ternary compound mimics a Li- or electron-doped binary hydride of MgH16. The parent hydride contains H2 molecules and is not a good superconductor. The extra electrons introduced break up the H2 molecules, increasing the amount of atomic hydrogen compared with the parent hydride, which is necessary for stabilizing the clathrate structure or other high-Tc structures. Our results provide a viable strategy for tuning the superconductivity of hydrogen-rich hydrides by donating electrons to hydrides via metal doping. Our approach may pave the way for finding high-Tc superconductors in a variety of ternary or quaternary hydrides. The research results have been published in Phys. Rev. Lett. 123, 097001 (2019) |
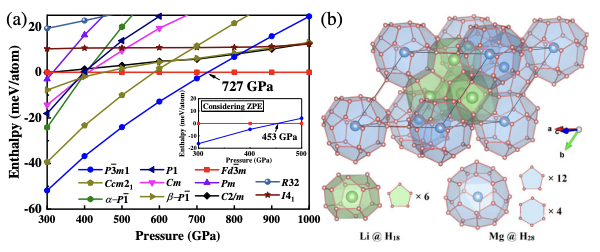 |
Polymeric nitrogen, stabilized by compressing pure molecular nitrogen, has yet to be recovered to ambient conditions, precluding its application as a high-energy density material. Here we suggest a route for synthesis of a tetragonal polymeric nitrogen, denoted t-N, via He-N compounds at high pressures. Using first-principles calculations with structure searching, we predict a class of nitrides with stoichiometry HeN4 that are energetically stable (relative to a mixture of solid He and N2) above 8.5 GPa. At high pressure, HeN4 comprises a polymeric channel-like nitrogen framework filled with linearly arranged helium atoms. The nitrogen framework persists to ambient pressure on decompression after removal of helium, forming pure polymeric nitrogen, t-N. t-N is dynamically and mechanically stable at ambient pressure with an estimated energy density of ~11.31 kJ/g, marking it out as a remarkable high-energy density material. This expands the known polymeric forms of nitrogen and indicates a route to its synthesis. The research results have been published in Phys. Rev. Lett. 119, 107001 (2017) |
 |
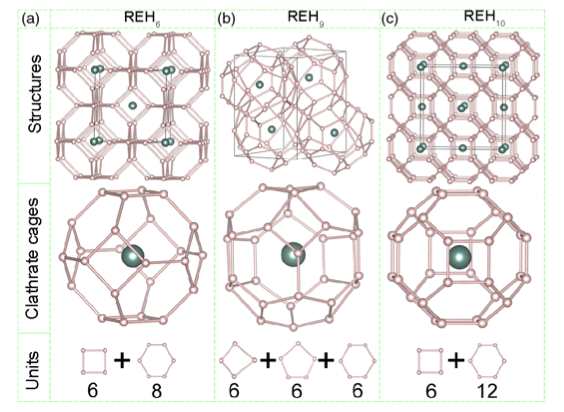
Room-temperature superconductivity has been a long-held dream and an area of intensive research. Recent experimental findings of superconductivity at 200 K in highly compressed hydrogen (H) sulfides have demonstrated the potential for achieving room-temperature superconductivity in compressed H-rich materials. We report first-principles structure searches for stable H-rich clathrate structures in rare earth hydrides at high pressures. The peculiarity of these structures lies in the emergence of unusual H cages with stoichiometries H24, H29, and H32, in which H atoms are weakly covalently bonded to one another, with rare earth atoms occupying the centers of the cages. We have found that high-temperature superconductivity is closely associated with H clathrate structures, with large H-derived electronic densities of states at the Fermi level and strong electron-phonon coupling related to the stretching and rocking motions of H atoms within the cages. Strikingly, a yttrium (Y) H32 clathrate structure of stoichiometry YH10 is predicted to be a potential room-temperature superconductor with an estimated 𝑇𝑐 of up to 303 K at 400 GPa, as derived by direct solution of the Eliashberg equation. The research results have been published in Nature Commun. 9, 722 (2018) |
 |
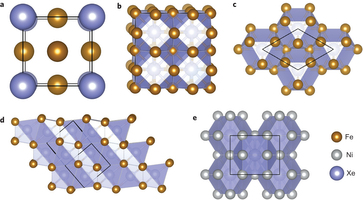
Studies of the Earth's atmosphere have shown that more than 90% of the expected amount of Xe is depleted, a finding often referred to as the 'missing Xe paradox'. Although several models for a Xe reservoir have been proposed, whether the missing Xe could be contained in the Earth's inner core has not yet been answered. The key to addressing this issue lies in the reactivity of Xe with Fe/Ni, the main constituents of the Earth's core. Here, we predict, through first-principles calculations and unbiased structure searching techniques, a chemical reaction of Xe with Fe/Ni at the temperatures and pressures found in the Earth's core. We find that, under these conditions, Xe and Fe/Ni can form intermetallic compounds, of which XeFe3 and XeNi3 are energetically the most stable. This shows that the Earth's inner core is a natural reservoir for Xe storage and provides a solution to the missing Xe paradox. The research results have been published in Nature Chem. 6, 644 (2014) |
 |
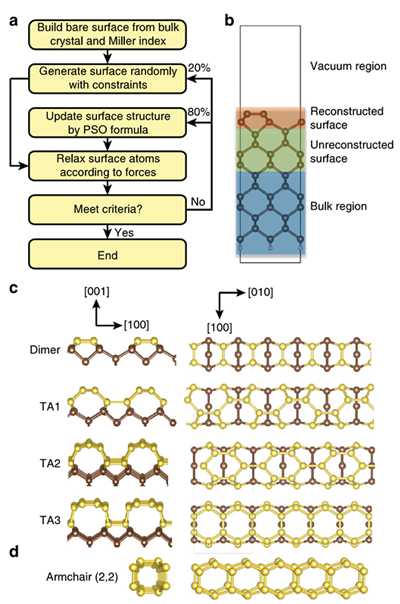
Surfaces of semiconductors are crucially important for electronics, especially when the devices are reduced to the nanoscale. However, surface structures are often elusive, impeding greatly the engineering of devices. Here we develop an efficient method that can automatically explore the surface structures using structure swarm intelligence. Its application to a simple diamond (100) surface reveals an unexpected surface reconstruction featuring self-assembled carbon nanotubes arrays. Such a surface is energetically competitive with the known dimer structure under normal conditions, but it becomes more favourable under a small compressive strain or at high temperatures. The intriguing covalent bonding between neighbouring tubes creates a unique feature of carrier kinetics (that is, one dimensionality of hole states, while two dimensionality of electron states) that could lead to novel design of superior electronics. Our findings highlight that the surface plays vital roles in the fabrication of nanodevices by being a functional part of them. The research results have been published in Nature Commun. 5, 3666 (2014). |
 |
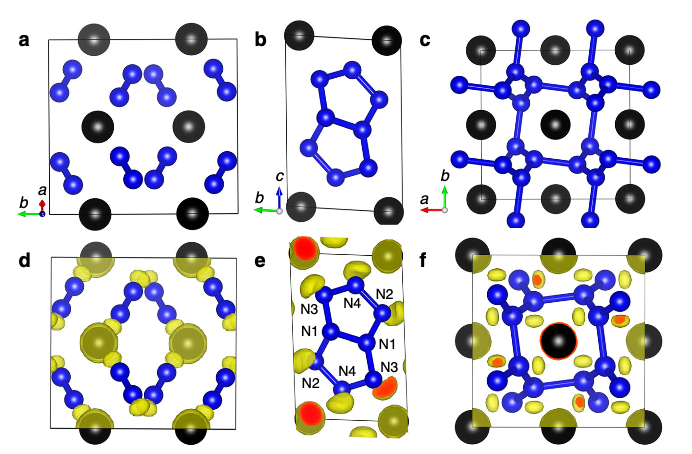
Carbon (C) is able to form various bonding patterns, including graphene sheets, chains and dimers, but stable bare six-membered C6 hexagonal rings, which are the fundamental structural motifs of graphite and graphene have long been missing. Here we report the stabilization of such bare C6 rings under high pressures in the charge-transfer systems of binary sesquicarbides Y2C3 and La2C3 as predicted by first-principles swarm structure searching simulations. We found that the external pressure can be used to efficiently tune structural transitions in the sesquicarbides from the ambient-pressure cubic phases into high-pressure orthorhombic phases, accompanied by significant C–C bonding modification from C–C dimers to bare C6 rings and polymerized graphene-like double C6 sheets. The bare C6 rings are stabilized in Y2C3 and La2C3 at pressures above 32 and 13 GPa, respectively, which are readily accessible to experiments. Chemical bonding analysis reveals that the bare C6 rings feature a benzene-like sp2 C–C bonding pattern with a delocalized π system. Y or La → C charge transfer and the need for denser structure packing are found to be part of the underlying mechanisms behind the stabilization of the bare C6 rings. The research results have been published in Chem. Sci. 5, 3936 (2014). |
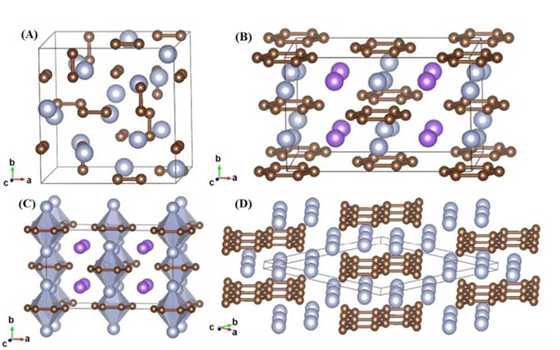 |
Tungsten borides are among a distinct class of transition-metal light-element compounds with superior mechanical properties that rival those of traditional superhard materials. An in-depth understanding of these compounds, however, has been impeded by uncertainties regarding their complex crystal structures. Here, we examine a wide range of chemical compositions of tungsten borides using a recently developed global structural optimization approach. We establish thermodynamically stable structures and identify a large number of metastable phases. These results clarify and correct previous structural assignments and predict new structures for possible synthesis. Our findings provide crucial insights for understanding the rich and complex crystal structures of tungsten borides, which have broad implications for further exploration of this class of promising materials. The research results have been published in Phys. Rev. Lett. 110, 136403 (2013). |
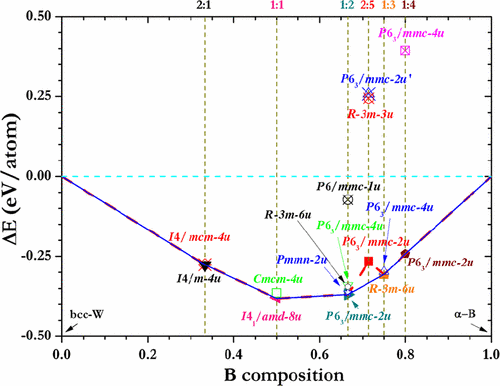 |
The recent discovery of phase IV of solid hydrogen and deuterium consisting of two alternate layers of graphenelike three-molecule rings and unbound H2 molecules have generated great interest. However, the vibrational nature of phase IV remains poorly understood. Here, we report a peculiar proton or deuteron transfer and a simultaneous rotation of three-molecule rings in graphenelike layers predicted by ab initio variable-cell molecular dynamics simulations for phase IV of solid hydrogen and deuterium at pressure ranges of 250–350 GPa and temperature range of 300–500 K. This proton or deuteron transfer is intimately related to the particular elongation of molecules in graphenelike layers, and it becomes more pronounced with increasing pressure at the course of larger elongation of molecules. As the consequence of proton transfer, hydrogen molecules in graphenelike layers are short lived and hydrogen vibration is strongly anharmonic. Our findings provide direct explanations on the observed abrupt increase of Raman width at the formation of phase IV and its large increase with pressure. The research results have been published in Phys. Rev. Lett. 110, 025903 (2013). |
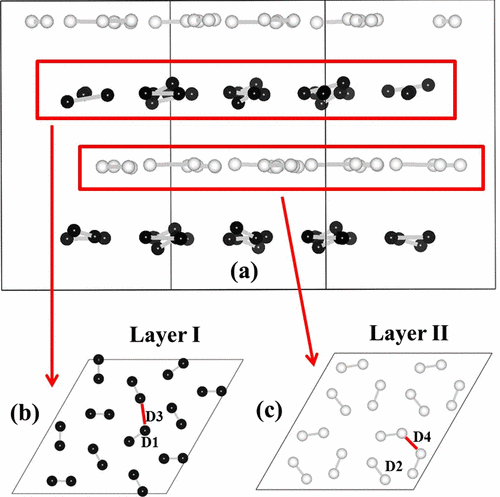 |
High pressure can fundamentally alter the bonding patterns of light elements and their compounds, leading to the unexpected formation of materials with unusual chemical and physical properties. Using an unbiased structure search method based on particle swarm optimization algorithms in combination with density functional theory calculations, we investigate the phase stabilities and structural changes of various Li-B systems on the Li-rich regime under high pressures. We identify the formation of four stoichiometric lithium borides (Li3B2, Li2B, Li4B, and Li6B) having unforeseen structural features that might be experimentally synthesizable over a wide range of pressures. Strikingly, it is found that the B-B bonding patterns of these lithium borides evolve from graphite-like sheets in turn to zigzag chains, dimers, and eventually isolated B ions with increasing Li content. These intriguing B-B bonding features are chemically rationalized by the elevated B anionic charges as a result of Li->B charge transfer. The research results have been published in J. Am. Chem. Soc. 134, 18699 (2012). |
 |
Under high pressure, triply bonded molecular nitrogen dissociates into singly bonded polymeric nitrogen, a potential high-energy-density material. The discovery of stable high-pressure forms of polymeric nitrogen is of great interest. We report the striking stabilization of cagelike diamondoid nitrogen at high pressures predicted by first-principles structural searches. The diamondoid structure of polymeric nitrogen has not been seen in any other elements, and it adopts a highly symmetric body-centered cubic structure with lattice sites occupied by diamondoids, each of which consists of ten nitrogen atoms, forming a N10 tetracyclic cage. Diamondoid nitrogen possesses a wide energy gap and is energetically most stable among all known polymeric structures above 263 GPa, a pressure that is accessible to a high-pressure experiment. Our findings represent a significant step toward the understanding of the behavior of solid nitrogen at extreme conditions. The research results have been published in Phys. Rev. Lett. 109, 175502 (2012). |
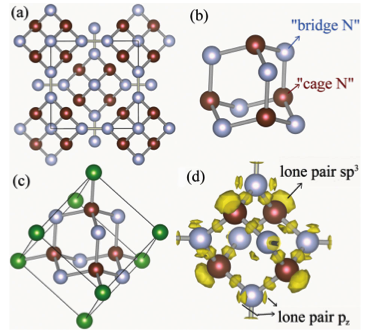 |
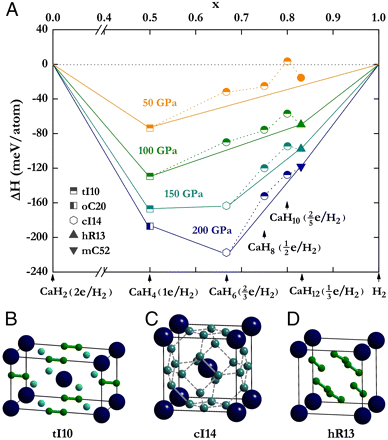
Hydrogen-rich compounds hold promise as high-temperature superconductors under high pressures. Recent theoretical hydride structures on achieving high-pressure superconductivity are composed mainly of H2 fragments. Through a systematic investigation of Ca hydrides with different hydrogen contents using particle swam optimization structural search, we show that in the stoichiometry CaH6 a body-centered cubic structure with hydrogen that forms unusual sodalite cages containing enclathrated Ca stabilizes above pressure 150 GPa. The stability of this structure is derived from the acceptance by two H2 of electrons donated by Ca forming an "H4" unit as the building block in the construction of the three-dimensional sodalite cage. This unique structure has a partial occupation of the degenerated orbitals at the zone center. The resultant dynamic Jahn-Teller effect helps to enhance electron-phonon coupling and leads to superconductivity of CaH6. A superconducting critical temperature (Tc) of 220-235 K at 150 GPa obtained from the solution of the Eliashberg equations is the highest among all hydrides studied thus far. The research results have been published in Proc. Natl. Acad. Sci. USA 109, 6463 (2012). |
 |
Oxygen is in many ways a unique element: It is the only known diatomic molecular magnet, and it exhibits an unusual O8 cluster in its high-pressure solid phase. Pressure-induced molecular dissociation as one of the fundamental problems in physical sciences has been reported from theoretical or experimental studies of diatomic solids H2, N2, F2, Cl2, Br2, and I2 but remains elusive for molecular oxygen. We report here the prediction of the dissociation of molecular oxygen into a polymeric spiral chain O4 structure (space group I41/acd, θ-O4) above 1.92-TPa pressure using the particle-swarm search method. The θ-O4 phase has a similar structure as the high-pressure phase III of sulfur. The molecular bonding in the insulating ε-O8 phase or the isostructural superconducting ζ-O8 phase remains remarkably stable over a large pressure range of 0.008-1.92 TPa. The pressure-induced softening of a transverse acoustic phonon mode at the zone boundary V point of O8 phase might be the ultimate driving force for the formation of θ-O4. Stabilization of θ-O4 turns oxygen from a superconductor into an insulator by opening a wide band gap (approximately 5.9 eV) that originates from the sp3-like hybridized orbitals of oxygen and the localization of valence electrons. The research results have been published in Proc. Natl. Acad. Sci. USA 109, 751 (2012). |
 |
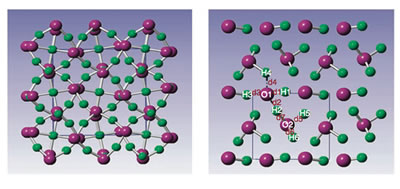
Water ice dissociates into a superionic solid at high temperature (>2,000 K) and pressure, where oxygen forms the lattice, but hydrogen diffuses completely. At low temperature, however, the dissociation into an ionic ice of hydronium (H3O)+ hydroxide (OH)- is not expected because of the extremely high energy cost (~1.5 eV) of proton transfer between H2O molecules. Here we show the pressure-induced formation of a partially ionic phase (monoclinic P21 structure) consisting of coupled alternate layers of (OH)δ- and (H3O)δ+ (δ=0.62) in water ice predicted by particle-swarm optimization structural search at zero temperature and pressures of >14 Mbar. The occurrence of this ionic phase follows the break-up of the typical O-H covalently bonded tetrahedrons in the hydrogen symmetric atomic phases and is originated from the volume reduction favourable for a denser structure packing. The research results have been published in Nature Commun. 2, 563 (2011). |
 |
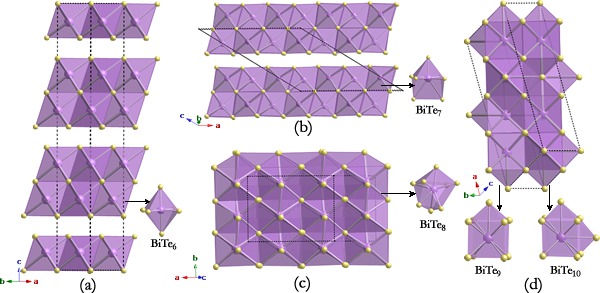
Being a best known thermoelectric material and a topological insulator at ambient condition, magic bismuth telluride (Bi2Te3) under pressure transforms into several superconducting phases, whose structures remain unsolved for decades. Here, we have solved the two long-puzzling low high-pressure phases as seven- and eightfold monoclinic structures, respectively, through particle-swarm optimization technique on crystal structure prediction. Above 14.4 GPa, we experimentally discovered that Bi2Te3 unexpectedly develops into a Bi-Te substitutional alloy by adopting a body-centered cubic disordered structure stable at least up to 52.1 GPa. The continuously monoclinic distortion leads to the ultimate formation of the Bi-Te alloy, which is attributed to the Bi-Te charge transfer under pressure. Our research provides a route to find alloys made of nonmetallic elements for a variety of applications. The research results have been published in Phys. Rev. Lett. 106, 145501 (2011). |
 |
Under high pressure, simple lithium (Li) exhibits complex structural behavior, and even experiences an unusual metal-to-semiconductor transition, leading to topics of interest in the structural polymorphs of dense Li. We here report two unexpected orthorhombic high-pressure structures Aba2-40 (40 atoms=cell, stable at 60-80 GPa) and Cmca-56 (56 atoms=cell, stable at 185-269 GPa), by using a newly developed particle swarm optimization technique on crystal structure prediction. The Aba2-40 having complex 4- and 8-atom layers stacked along the b axis is a semiconductor with a pronounced band gap >0.8 eV at 70 GPa originating from the core expulsion and localization of valence electrons in the voids of a crystal. We predict that a local trigonal planar structural motif adopted by Cmca-56 exists in a wide pressure range of 85??C434 GPa, favorable for the weak metallicity. The research results have been published in Phys. Rev. Lett. 106, 015503 (2011). |
 |
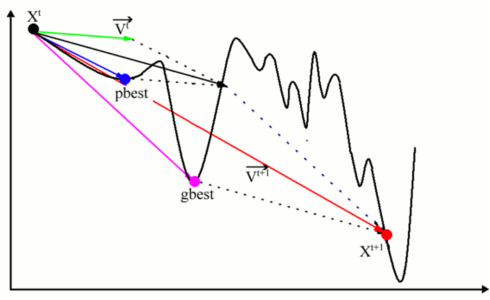 |
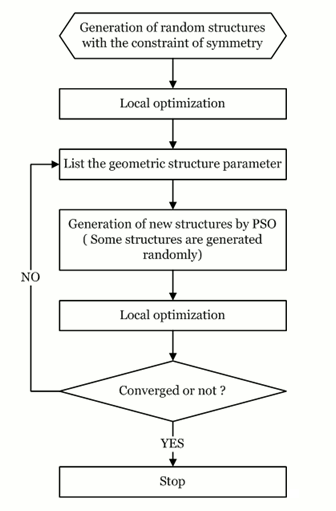
We have developed a method for crystal structure prediction from ?0?3??scratch?0?3?? through particle swarm optimization (PSO) algorithm within the evolutionary scheme. PSO technique is different with the genetic algorithm and has apparently avoided the use of evolution operators (e.g., crossover and mutation). The approach is based on an efficient global minimization of free energy surfaces merging total-energy calculations via PSO technique and requires only chemical compositions for a given compound to predict stable or metastable structures at given external conditions (e.g., pressure). A particularly devised geometrical structure parameter which allows the elimination of similar structures during structure evolution was implemented to enhance the structure search efficiency. The application of designed variable unit cell size technique has greatly reduced the computational cost. Moreover, the symmetry constraint imposed in the structure generation enables the realization of diverse structures, leads to significantly reduced search space and optimization variables, and thus fastens the global structure convergence. The PSO algorithm has been successfully applied to the prediction of many known systems (e.g., elemental, binary and ternary compounds) with various chemical bonding environments (e.g., metallic, ionic, and covalent bonding). The high success rate demonstrates the reliability of this methodology and illustrates the promise of PSO as a major technique on crystal structure determination. The research results have been published in Phys. Rev. B 82, 094116 (2010). |
 |